Машинное обучение в химии: как найти кандидата в лекарство за дни вместо 15 лет
Фармацевтическая разработка лекарств занимает от десяти до пятнадцати лет и обходится в миллиарды долларов, но машинное обучение в химии позволяет сократить путь от идеи до кандидата в лекарства до нескольких дней работы за ноутбуком.

Весь рабочий процесс, от поиска биологической мишени до генерации новых молекул, строится на бесплатных открытых инструментах и запускается в Google Colab без специализированного оборудования: российским хемоинформатикам и разработчикам лекарств не нужен бюджет фармкорпорации, чтобы получить первых кандидатов на ингибиторы (вещества, которые блокируют работу белка-мишени).
Туториал воспроизводит полный цикл поиска молекул-кандидатов для подавления мутантного рецептора EGFR (рецептор эпидермального фактора роста, белок на поверхности раковых клеток, который заставляет их бесконтрольно делиться). Мутация C797S делает опухоль устойчивой к осимертинибу, препарату третьего поколения. Задача: научить модель на известных данных о молекулах-ингибиторах EGFR и предложить новые, не описанные ранее аналоги как стартовые точки для лекарства четвёртого поколения.
Машинное обучение в химии здесь работает по принципу QSAR (количественная связь «структура-активность»): алгоритм находит закономерности между строением молекулы и её биологической активностью, а затем использует эти закономерности для оценки новых соединений.
Что понадобится
- Google Colab (бесплатный аккаунт) или локальный Python 3.9+
- Библиотеки: RDKit (работа с молекулами), scikit-learn (машинное обучение), SHAP (объяснение предсказаний модели), matplotlib (графики), requests (доступ к API)
- Открытые базы данных: ChEMBL (база биоактивности), UniProt (данные о белках), PubChem (проверка новизны молекул)
- Время: 2 до 4 часов на первый полный прогон с разбором результатов
- Знания: базовый Python, общее понимание органической химии (подробные пояснения даны по ходу шагов)
Пошаговая инструкция
- Установите зависимости и настройте среду. В Colab или терминале выполните установку пакетов. Код ниже проверяет наличие каждой библиотеки и доустанавливает недостающие:
import subprocess, sys
for pkg in ["rdkit", "shap", "requests"]:
subprocess.run([sys.executable, "-m", "pip", "install", "-q", pkg], check=False)
Импортируйте основные модули: numpy, pandas, RDKit, sklearn, matplotlib, SHAP. Зафиксируйте случайное зерно (random seed = 42), чтобы результаты воспроизводились.
- Определите биологическую мишень. Через API ChEMBL найдите запись EGFR человека (идентификатор CHEMBL203). Скрипт автоматически выбирает мишень с наибольшим числом записей биоактивности IC50. Дополнительно подтяните описание функции белка из UniProt по идентификатору accession.
TARGET_CHEMBL_ID = "CHEMBL203"
# запрос к ChEMBL API:
url = "https://www.ebi.ac.uk/chembl/api/data/target/CHEMBL203"
-
Извлеките данные биоактивности из ChEMBL. Запросите все записи IC50 с указанным pChEMBL value (логарифмическая мера активности: чем выше значение, тем сильнее молекула подавляет мишень). Ограничьте выборку до 9 000 записей и до 4 000 уникальных молекул, чтобы обучение проходило на рабочей машине.
-
Очистите и стандартизируйте молекулы. С помощью RDKit:
- удалите соли и противоионы из записей
- стандартизируйте структуры (канонические SMILES, линейная запись структуры молекулы)
-
объедините повторные измерения (медиана pIC50 для одной молекулы)
-
Вычислите молекулярные дескрипторы. Для каждой молекулы рассчитайте:
- Отпечатки Моргана (Morgan fingerprints, 2048 бит, радиус 2) : бинарное представление фрагментов структуры
- Физико-химические свойства: молекулярная масса, логарифм липофильности (logP), число доноров и акцепторов водородных связей
-
Проанализируйте разнообразие скаффолдов (скаффолд, «скелет» молекулы по Murcko)
-
Разделите данные по скаффолдам и обучите модель. Scaffold-split (разбиение, при котором молекулы с одинаковым скелетом попадают только в одну выборку) гарантирует, что модель проверяется на структурно новых соединениях. Обучите Random Forest (случайный лес) на тренировочной выборке:
from sklearn.ensemble import RandomForestRegressor
model = RandomForestRegressor(n_estimators=500, random_state=42)
model.fit(X_train, y_train)
Оцените качество по R², RMSE и коэффициенту корреляции Спирмена на тестовой выборке.
-
Интерпретируйте модель через SHAP. Библиотека SHAP (SHapley Additive exPlanations) показывает, какие именно фрагменты молекулы толкают предсказание активности вверх или вниз. Визуализируйте субструктуры, которые модель считает важными для подавления EGFR.
-
Сгенерируйте новые молекулы-кандидаты. Метод BRICS (разрезание молекулы по синтетически реалистичным связям) разбивает активные молекулы на фрагменты. Скрипт рекомбинирует фрагменты от 60 наиболее активных «родителей», выполняя до 4 000 попыток сборки. Каждый результат проходит фильтры:
- предсказанная pIC50 выше 7.0 (граница активности)
- соответствие критериям drug-likeness (пригодности к разработке как лекарства)
- оценка синтезируемости (насколько реально синтезировать молекулу в лаборатории)
-
проверка новизны: не совпадает ли со структурами в PubChem
-
Отберите финальный шортлист. Из прошедших все фильтры молекул выберите 12 лучших кандидатов, ранжированных по комбинированному баллу «активность + пригодность к разработке». Визуализируйте их структуры и сохраните таблицу для передачи медицинским химикам.
На входе: идентификатор мишени CHEMBL203 (EGFR) и параметры модели (500 деревьев, отпечатки 2048 бит). На выходе после полного прогона: таблица из 12 молекул-кандидатов с предсказанной pIC50 выше 7.0, оценками QED (drug-likeness), SA Score (синтезируемость) и подтверждением, что структура отсутствует в PubChem. Каждая молекула визуализирована с подсветкой фрагментов, которые SHAP отметил как значимые для активности. Полный ноутбук занимает около 300 строк кода и выполняется в Google Colab за 15 до 40 минут в зависимости от скорости API.
- Случайное разбиение вместо scaffold-split. Если тренировочная и тестовая выборки содержат молекулы с одинаковым скелетом, метрики раздуваются: модель «запоминает» скаффолд, а не учится обобщать. Всегда используйте scaffold-split для QSAR.
- Игнорирование агрегации повторов. Одна молекула может иметь 5 до 10 измерений IC50 из разных лабораторий. Без усреднения (медианы) модель получает противоречивые метки и учится на шуме.
- Доверие к предсказанной pIC50 без экспериментальной проверки. Модель QSAR прогнозирует, но не доказывает активность. Каждый кандидат требует синтеза и биологического тестирования.
- Пропуск проверки новизны. Без сопоставления с PubChem вы рискуете «открыть» уже известное вещество или запатентованную структуру.
- Ошибки при стандартизации SMILES. Соли, таутомеры (разные формы записи одной молекулы), нестандартные заряды искажают отпечатки. Всегда пропускайте входные данные через rdMolStandardize.
Что делать с этим прямо сейчас?
Хемоинформатику и разработчику лекарств в РФ: весь процесс запускается бесплатно и легально. ChEMBL, UniProt и PubChem доступны без ограничений по региону. Замените CHEMBL203 на идентификатор вашей мишени и повторите пайплайн для собственного проекта.
Автору Дзена, который пишет о науке и технологиях: сам факт, что полный цикл drug discovery укладывается в один Colab-ноутбук, отличная основа для разбора или серии постов. Покажите скриншоты визуализации SHAP: они наглядно объясняют, «почему модель считает эту молекулу активной».
Предпринимателю в биотехе: если в команде есть хотя бы один Python-разработчик с базовым пониманием химии, этот workflow заменяет первичный скрининг, на который в CRO (контрактная исследовательская организация) уходят недели и сотни тысяч рублей. Результат не замена полного цикла R&D, но качественный входной фильтр.
По моим наблюдениям, главная ценность подобных открытых пайплайнов не в том, что они выдают готовое лекарство. Они снижают порог входа: аспирант в Томске или Казани получает тот же инструментарий, что и группа в Бостоне. Честная оговорка: от кандидата в ноутбуке до реальной молекулы в клинике по-прежнему лежат годы мокрой химии, доклинических испытаний и регуляторных процедур. Модель Random Forest на отпечатках Моргана не учитывает трёхмерную структуру белка и динамику связывания. Для серьёзного проекта стоит дополнить пайплайн докингом (моделирование посадки молекулы в активный сайт белка) и молекулярной динамикой. Но как первый фильтр и обучающий проект это рабочий инструмент, который стоит попробовать.
Научитесь использовать ИИ для своих задач
В dzen.guru мы разбираем практические сценарии применения нейросетей от контента до науки. Подпишитесь, чтобы не пропустить следующие гайды.
Подписаться на dzen.guruДвенадцать молекул в таблице не вылечат рак, но они делают кое-что важнее: доказывают, что первый этап поиска лекарства больше не заперт за дверью с бюджетом в миллиард.

Основатель dzen.guru. Эксперт по монетизации и продвижению на Дзен. Автор курса «Старт на Дзен 2026».
Читайте также
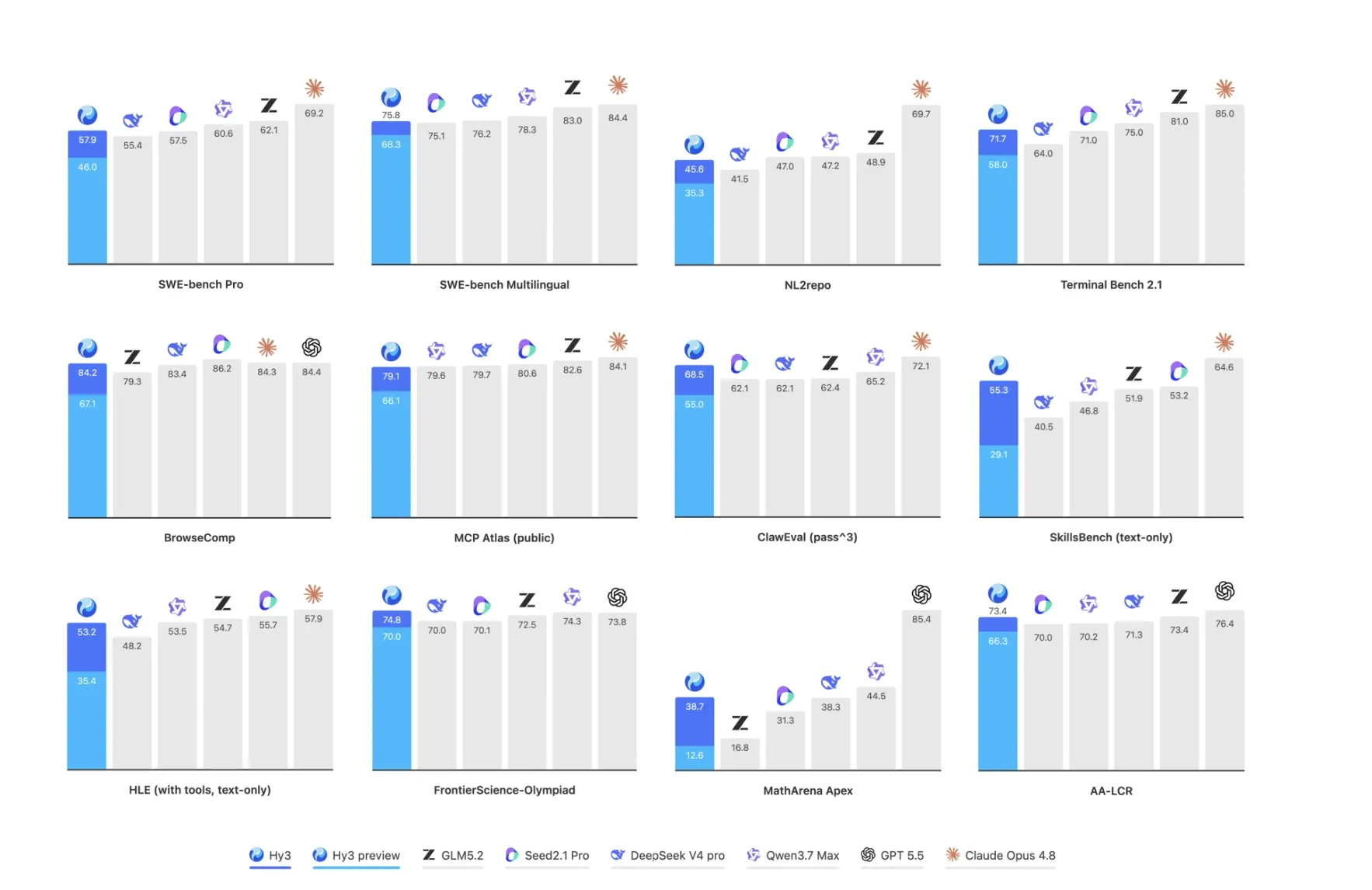
Tencent открыла Hy3: mixture of experts модель на 295B параметров бесплатна до 2026 года
Tencent второго июля выпустила Hy3, открытую модель на 295 миллиардов параметров с архитектурой MoE (mixture of experts, «смесь экспертов», когда из сотен…

Что такое ИИ-агент: кейс Vercel, где 3 млн развёртываний в день идут без людей
Чтобы понять, что такое ИИ-агент (AI agent, программа, которая сама выбирает инструменты и выполняет цепочку действий для достижения цели), достаточно…
OpenAI выпустила GPT Realtime 2.1: голосовые агенты рассуждают вслух втрое дешевле
OpenAI 12 июня выпустила два голосовых ИИ-агента нового поколения, и младшая модель впервые умеет рассуждать в реальном времени, отвечая голосом втрое дешевле…
Комментарии